Research Overview
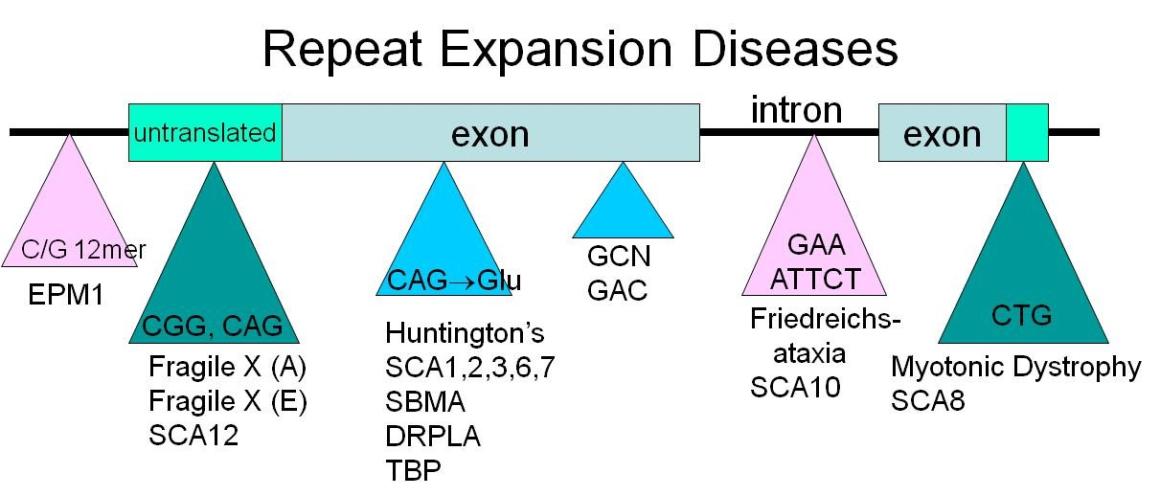
The overall goal of my research is to understand mechanisms of genome stability that operate at repetitive DNA, and how they collaborate to protect against repeat expansions and chromosome fragility. These are important questions, as they have direct bearing on human diseases, both repeat expansion diseases and cancer, and therefore human health.
A primary aim of my lab is to elucidate the mechanisms involved in repeat instability and fragility, and determine how these two unusual characteristics are interrelated. We have focused most of our research on the expandable CAG/CTG repeat, but have also studied other types of repeats that expand to cause human disease.
A second major focus of the lab is to understand how repetitive DNA causes genomic instability, including DNA breaks and fork stalling, and the cellular mechanisms used to overcome these barriers to genome integrity. We use a combination of genetic and physical assays to study the types of proteins that mediate replication of structure-forming repeats and repair of repeat-induced chromosomal damage. It is important to understand the mechanisms that are utilized by the cell to maintain genomic integrity in the face of structure-forming DNA sequences like the expandable triplet repeats, and they present a powerful model for obtaining unique insights into genome protective mechanisms.
Some of the past contributions of the lab and ongoing projects are listed below.
Identification of the role of key cellular enzymes needed to protect against repeat expansions
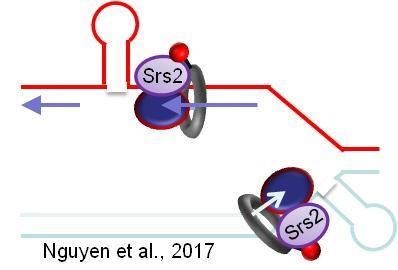
We have identified multiple proteins and pathways needed to maintain repeat stability through directed testing as well as novel genetic assays to screen for proteins whose expression influences repeat expansion or fragility. Examples include the FEN1/Rad27 flap endonuclease, the Srs2 helicase and its human ortholog RTEL, and the alternative PCNA clamp loader Ctf18-RFC . We also discovered that expanded repeats activate cellular S and G2 checkpoints, and that the checkpoint response is important for preventing both repeat fragility and instability. We identified homologous recombination as being both a key protective mechanism to heal breaks that occur at expanded CAG repeats, as well as a mechanism that can cause repeat expansions. We are actively studying pathways of DNA repair that impact repeat instability and their mechanism of action.
The role of chromatin structure and chromatin modifications in maintaining CAG repeats
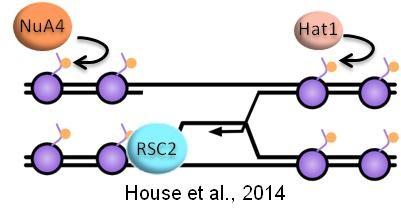
DNA repair takes place in the context of chromatin structure, and CAG/CTG repeat sequence is an especially strong nucleosome positioning sequence. We have identified several histone modifications and chromatin remodeling enzymes that are particularly important in facilitating DNA transactions in the context of CAG repeats. Recently, we identified acetylation of the histone H4 tail and the RSC remodeler as being especially important for facilitating sister chromatid recombination and preventing repeat expansions from occurring during this process. We are investigating how these and other histone modifications and chromatin remodelers facilitate genome stability and promote repair with fidelity in the context of structure-forming DNA repeats.
Replication of structure-forming repeats and the role of fork restart in preventing chromosome breaks and repeat instability
The Freudenreich lab is interested in how replication proceeds through repetitive DNA tracts, identifying the Srs2 helicase as being particularly important for replication through hairpin structures, and characterizing the role of fork stabilizing complexes and fork restart in maintaining CAG repeats.
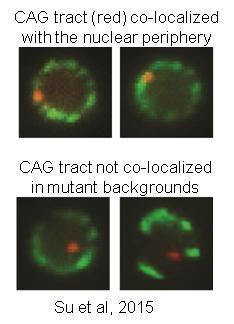
Recently, we identified S-phase re-localization to the nuclear pore as a key protective mechanism that prevents fragility and instability of expanded CAG repeats. Our data suggests that localization to the nuclear pore occurs due to replication fork collapse, and that it impacts fork restart and DNA repair at expanded triplet repeats. We are actively investigating the mechanisms involved in this fascinating process.
The molecular basis of common fragile sites
In addition to triplet repeats, several other types of microsatellite sequences act as "common" fragile sites on human chromosomes - sites present in all individuals that are prone to breakage. The causes of chromosome fragility are not well understood, but the results can be significant: a broken chromosome is vulnerable to loss or rearrangement, which can lead to cancer or other genetic diseases.
My lab is interested in the basis for chromosome fragility at common fragile sites, which are often found deleted or rearranged in cancer cells. We discovered that a sequence within the human site FRA16D containing a polymorphic AT repeat can cause fragility on a yeast chromosome, and can stall replication forks. Interference with replication fork progression correlated with the potential to form a hairpin or cruciform DNA structure, suggesting that interference with replication by structure-forming sequences could contribute to the fragility of the FRA16D region.
We are currently studying both sequence elements and protein requirements for breakage at common fragile site FRA16D, and are testing whether the conditions and proteins that affect triplet repeats also affect these sequences. The results could shed light on the genomic instability that occurs during tumorigenesis.